Scientists in Dr. Hongbing Wang’s lab are currently focused on three major research initiatives.
Regulation of Drug-metabolizing Enzymes by Xenobiotic Receptors
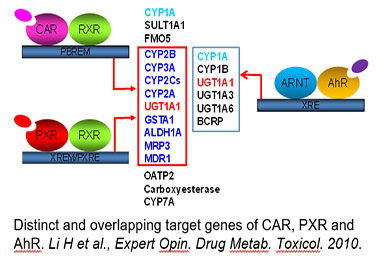
Drug-metabolizing enzymes (DMEs) and transporters play pivotal roles in the disposition and detoxification of numerous endogenous and xenobiotic chemicals. To accommodate chemical challenges, the expression of many DMEs and transporters are up-regulated by a group of ligand-activated transcription factors. The importance of such transcription factors in xenobiotic metabolism and clearance is best exemplified by the promiscuous xenobiotic receptors: the pregnane X receptor (PXR, NR1I2), constitutive androstane receptor (CAR, NR1I3), and aryl hydrocarbon receptor (AhR). Together, these receptors govern the inductive expression of a large array of target genes encoding phase I and II DMEs, and drug transporters. Moreover, these receptors also exhibit distinctive mechanisms of activation: ligand-dependent (direct) and ligand-independent (indirect) activation. Over the years, we have investigated the transcriptional regulation of CYP2B6, CYP3A4, BCRP, and SLC13A5 in human liver by different xenobiotic receptors, with CYP2B6 as a primary target used to illustrate the mechanisms and contribution of CAR and PXR in hepatic P450 induction. We have shown that in addition to CAR and PXR, other liver-enriched transcriptional factors such as HNF4α, C/EBPα, and HNF3β also contribute to the optimal transcription of drug-metabolizing enzymes in the liver.
Dual activation of CAR and Nrf2 in Cyclophosphamide/doxorubicin-based Chemotherapy
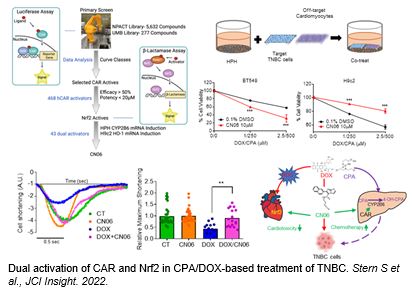 Cyclophosphamide (CPA) and doxorubicin (DOX) are key components of chemotherapy for triple-negative breast cancer (TNBC) although suboptimal outcomes are commonly associated with drug resistance and/or intolerable side-effects. Through an approach combining high-throughput screening and chemical modification, we developed CN06 as a dual activator of the constitutive androstane receptor (CAR) and nuclear factor erythroid 2-related factor 2 (Nrf2). CN06 enhances CAR-induced bioactivation of CPA (a prodrug) by provoking hepatic expression of CYP2B6, while repressing DOX-induced cytotoxicity in cardiomyocytes in vitro via stimulating Nrf2-antioxidant signaling. Utilizing a multicellular co-culture model incorporating human primary hepatocytes, TNBC cells, and cardiomyocytes, we show that CN06 increased CPA/DOX-mediated TNBC cell death via CAR-dependent CYP2B6 induction and subsequent conversion of CPA to its active metabolite 4-hydroxy-CPA, while protecting against DOX-induced cardiotoxicity by selectively activating Nrf2-antioxidant signaling in cardiomyocytes but not in TNBC cells. Further, CN06 preserves the viability and function of human iPSC-derived cardiomyocytes by modulating antioxidant defenses, decreasing apoptosis, and enhancing the kinetics of contraction and relaxation. Collectively, our findings identify CAR and Nrf2 as novel combined therapeutic targets whereby CN06 holds the potential to improve the efficacy:toxicity ratio of CPA/DOX-containing chemotherapy.
Cyclophosphamide (CPA) and doxorubicin (DOX) are key components of chemotherapy for triple-negative breast cancer (TNBC) although suboptimal outcomes are commonly associated with drug resistance and/or intolerable side-effects. Through an approach combining high-throughput screening and chemical modification, we developed CN06 as a dual activator of the constitutive androstane receptor (CAR) and nuclear factor erythroid 2-related factor 2 (Nrf2). CN06 enhances CAR-induced bioactivation of CPA (a prodrug) by provoking hepatic expression of CYP2B6, while repressing DOX-induced cytotoxicity in cardiomyocytes in vitro via stimulating Nrf2-antioxidant signaling. Utilizing a multicellular co-culture model incorporating human primary hepatocytes, TNBC cells, and cardiomyocytes, we show that CN06 increased CPA/DOX-mediated TNBC cell death via CAR-dependent CYP2B6 induction and subsequent conversion of CPA to its active metabolite 4-hydroxy-CPA, while protecting against DOX-induced cardiotoxicity by selectively activating Nrf2-antioxidant signaling in cardiomyocytes but not in TNBC cells. Further, CN06 preserves the viability and function of human iPSC-derived cardiomyocytes by modulating antioxidant defenses, decreasing apoptosis, and enhancing the kinetics of contraction and relaxation. Collectively, our findings identify CAR and Nrf2 as novel combined therapeutic targets whereby CN06 holds the potential to improve the efficacy:toxicity ratio of CPA/DOX-containing chemotherapy.
Function and Regulation of SLC13A5 in the Liver
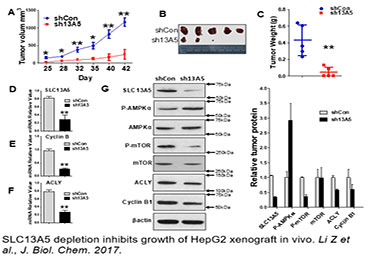
Citrate is a key energy sensor that plays a central role in carbohydrate metabolism, energy production, and histone acetylation. The intracellular level of citrate is tightly controlled through a balance of biosynthesis and transport. In the liver, the solute carrier family 13 member 5 (SLC13A5), a sodium-coupled citrate transporter, is essential for the import of citrate from the circulation to hepatocytes, a process that can be perturbed by both xenobiotic and endobiotic stimuli. Recent studies have shown that expression of SLC13A5 was increased in obese, non-alcoholic fatty liver disease (NAFLD) patients, high-fat diet (HFD)-treated rhesus monkeys, and in xenobiotic-treated human and rat hepatocytes, suggesting upregulation of SLC13A5 can be a risk factor for metabolic disorders. However, despite the emerging importance of SLC13A5 in energy homeostasis, the mechanism(s) by which the SLC13A5 gene is transcriptionally regulated and whether clinically used drugs disturb the expression of this transporter are not well characterized. Moreover, whether SLC13A5 affects hepatic functions beyond lipid homeostasis is largely unknown. To this end, we have shown that 1) prototypical activators of the constitutive androstane receptor (CAR) and the pregnane X receptor (PXR) robustly induce expression of human SLC13A5; 2) knockdown of SLC13A5 attenuates the proliferation of hepatocellular carcinoma cells; and 3) expression of SLC13A5 is inversely correlated with the activation of AMPK signaling. Building on these findings, we expect that further studies will yield novel knowledge regarding the transcriptional regulation of SLC13A5 in the liver, and offer experimental evidence that targeted disruption of SLC13A5 as a nutrient regulator will alter the proliferation of hepatoma cells by modulating AMPK/mTOR signaling pathways.
Novel noncanonical Actions of CAR in Human Liver
CAR is a well-established xenobiotic sensor that regulates the expression of numerous genes encoding proteins important for drug metabolism and clearance. Accumulating evidence suggests that CAR also plays noncanonical roles in coordinating diverse physiological and pathophysiological responses associated with energy homeostasis and cell proliferation. Studies in rodents have established activation of CAR as a key event promoting liver tumor formation. In contrast, CAR activation-induced replicative DNA synthesis and hepatocyte proliferation in rodents were not observed in either cultured human liver cells in vitro or in chimeric mice with humanized liver in vivo. Moreover, epidemiological studies have shown that even after long-term clinical use, phenobarbital, a prototypical CAR activator, does not increase the incidence of liver tumors in humans. The role of human CAR (hCAR) in hepatoma cell proliferation and liver cancer development remains poorly understood. The aim of this project is to delineate the role of hCAR in liver tumor progression and to develop a comprehensive understanding of the molecular mechanisms underlying the effects of hCAR on hepatoma cell proliferation. We have shown that 1) expression of hCAR was significantly lower in hepatocellular carcinoma (HCC) compared to normal liver and, importantly, hCAR expression is inversely correlated with HCC outcomes; 2) ectopic expression of the reference hCAR but not a splicing variant isoform (hCAR3) in hepatoma cells markedly repressed cell proliferation, soft agar colony formation, and the growth of hepatoma xenografts in nude mice; 3) RNA-seq analyses revealed that hCAR alters the expression of a cluster of tumor suppressors and oncogenes including the downregulation of erythropoietin (EPO), a pleiotropic growth factor that exhibits cell proliferation and anti-apoptosis functions; and 4) activation of human and mouse CAR differentially alters the expression of cell proliferation genes in vivo. We expect to determine the role of hCAR in HCC development and provide novel mechanistic insights into hCAR-mediated suppression of HCC progression that will open the door to novel biomarkers and therapeutics.